X-ray, Electron and NMR Crystallography to Structural Determination of Small Organic Molecules
JEOL NEWS Vol.53 No.6
Yusuke Nishiyama1, 2
1 JEOL RESONANCE Inc. 2 RIKEN CLST-JEOL Collaboration Center
Here we present the recent development at RIKEN CLST-JEOL collaboration laboratory to explore the molecular structures of low-molecular weight pharmaceutical compounds in natural abundance (without any isotopic labeling); the recent progress in fast magic angle spinning (MAS) technology in solid-state nuclear magnetic resonance (ssNMR) and in ultra high sensitivity camera in transmission electron microscopy (TEM) paves a new way to answer problems in the pharmaceutical industry and sciences. 1) Crystalline polymorphs and 2) salt/cocrystal are two major concerns in terms of quality control, stability, and intellectual property. To identify the crystalline form, powder X-ray diffraction and 13C cross-polarization MAS ssNMR are widely used methods, however, the former is sometimes not suitable for mixture analysis and latter fails to distinguish crystalline forms with similar molecular conformations. To solve these issues, we use electron diffraction (ED) and 1H fast MAS NMR. The crystalline form can be determined from nano- to micro-meter sized single crystals using ED, since electron interactions are 4 to 5 order stronger than X-ray interactions. 1H NMR also gives suitable information to molecular packing since 1H is located at the surface of the crystals. The salt/cocrystal issue, where hydrogen plays a key role, is a serious problem, since single crystal X-ray diffraction (SCXRD) cannot determine the hydrogen atom position precisely. Here we determine the internuclear distances between 1H and 15N using ssNMR at fast MAS conditions, while the global structure is obtained through SCXRD, answering the salt/cocrystal questions.
Introduction
While biotechnology-based medicines are listed at the top shares in pharmaceutical market, the traditional low-molecular weight drugs are still very important for daily treatment, for example adult diseases. These low-molecular weight active pharmaceutical ingredients (APIs) can typically be crystallized in several different forms, i.e. crystalline polymorphs, depending on the crystallization conditions. Since the solubility and stability are largely affected by the crystalline form, it is very important to control and monitor the crystalline form in view point of quality control from development to production stages [1, 2, 3]. When the crystals with large enough size (~100 μm) is available, single crystal X-ray diffraction (SCXRD) gives distinct answer to crystalline forms with atomic resolution. However, most of the low-molecular weight drugs are provided in micro-crystalline forms with various formulation including tablets, pills, powders. As these formulations involve the excipient, it is crucial to determine crystalline form from micro-crystals in mixture. Powder X-ray diffraction (PXRD) and 13C cross-polarization magic angle spinning nuclear magnetic resonance (CPMAS NMR) are two major methods to identify the crystalline form. As the experimental patterns/spectra gives finger prints of each crystalline form, the comparison of patterns/spectra between drug and standard form gives, in most cases, clear answer to the crystalline from in it. However, both methods still have practical problems. PXRD sometimes fails to identify the crystalline form as many diffraction patters from API and excipient are overlapped to each other. On the other hand, 13C CPMAS which is sensitive to molecular conformation is suitable method for mixture analysis as most signals from excipient appear at the positions different from those from API, avoiding the overlapping of signals. However, 13C CPMAS is rather insensitive to molecular packing as carbon atoms are buried inside the molecules and located far from the molecular surface. Thus, the 13C CPMAS fails to identify the crystalline forms with similar molecular conformations. Moreover, the sensitivity to crystalline form is less than PXRD.
The other major issue in low-molecular weight API is way to identify salt/cocrystal/continuum in multi-component systems. It is sometimes required to improve the solubility and/or stability of API crystals. The multi-component systems composed with API and inert coformer are one of the widely used solutions to this issue. Many examples can be found in multi-component systems by mixing the (typically basic) API and (acidic) coformer. When the difference of pKa (ΔpKa) is larger than 3, this forms salt, where intermolecular ionic interactions are found. In salt, proton from coformer totally moves toward API. Recently, another class of multi-component systems, cocrystal, is introduced when ΔpKa is less than 3. In cocrystal, the proton in coformer still stays there and intermolecular hydrogen bonding is formed. In addition, the system between salt and cocrystal is also found, i.e. continuum where proton is located between API and coformer. It is crucial to identify salt/cocrystal/continuum in terms of intellectual view point, especially when ΔpKa is smaller than 3. However, lack of ability to determine the proton position in XRD based methods brings serious issue to the identification of salt/cocrystal/continuum, since the difference between them comes only from hydrogen positions.
The major issues which are not solved yet are 1) analysis of mixture, 2) identification of crystalline form with similar molecular conformation, and 3) lack of ability to determine the 1H positions. The former two issues are related to crystalline polymorphs and the last one is to salt/cocrystal/continuum. Here, we combine electron diffraction (ED), 1H ssNMR, and SCXRD to tackle with these issues. ED is one of the observation methods in transmission electron microscopy (TEM) equipments and gives diffraction patterns. Since interaction of electron is 104 to 105 stronger than that of X-ray, ED pattern can be observed from nano- to micro-sized single crystals, enabling mixture analysis. Although the application of ED has largely been limited to inorganic materials which are robust to electron irradiation, recent progress in high-sensitivity camera allows low-dose mode together with cryo sample holder, paving a way to ED observation of organic crystals including low-molecular weight API. The issue 2) is raised because 13C CPMAS is insensitive to molecular conformation as mentioned above. Fortunately, the introduction of very fast MAS technology > 70 kHz enables the high resolution 1H observation even in rigid solids [4, 5, 6], where 1Hs are tightly coupled with 1H-1H dipolar interactions at moderate MAS rate. Since 1H is located at the surface, 1H isotropic chemical shifts are sensitive to not only conformation but also molecular packings. In addition, 1H-1H intermolecular correlation gives distinct patterns for each molecular packing. The fast MAS technology realizes not only high resolution 1H NMR but also several sophisticated experiments, including 14N NMR and 1H-15N distance measurements. The former reveals protonated state in small organic molecules. The latter gives clear answer to salt/cocrystal/continuum issue where hydrogen positions play a key role. In addition, we evaluate the throughput of each method.
Crystalline Polymorphs[7]
Identification of crystalline forms in pharmaceutical applications is crucial and usually performed by PXRD and 13C CPMAS ssNMR. However, the former is not suitable for mixture analysis and the latter sometimes fails as discussed above. As an example, 13C CPMAS spectra of three different (pseudo) polymorphs of L-histidine are shown in Fig. 1. L-histidine (Fig. 1b, c) gives distinct spectra from its hydrochloride salt of L-histidine·HCl·H2O (Fig. 1a), because of different molecular conformation. On the other hand, L-histidine in orthorhombic (Fig. 1b) and monoclinic (Fig. 1c) form gives almost identical 13C CPMAS spectra, reflecting the close conformation to each other. As shown here, 13C CPMAS is sensitive measure to identify crystalline form with different conformation, however, fails to distinguish the polymorphs with similar molecular conformation. We here propose a combined approach of ED and 1H ssNMR at very fast MAS to answer these questions.
Figure 2 shows TEM image and ED pattern of L-histidine·HCl·H2O measured at room temperature. While the size of crystal is about 1 μm, which is typically found in pharmaceutical products, 100 nm nano-beam is applied. Thus, in principle, crystals with 100 nm are large enough to obtain single crystalline ED patterns. In fact, we have measure ED patterns from 100 nm or even smaller crystals. As the critical dose of L-histidine·HCl·H2O at room temperature is only 10-20 e-nm-2, we set the dose rate to 10 e-nm-2s-1 with exposure time of 1 s, resulting in a total dose of 10 e-nm-2. Even with such a very low dose conditions, the state-of-the-art CMOS camera (OneView, Gatan, Inc., USA) gives enough sensitivity. While the critical doses of 10-20 e-nm-2 are typically found in organic molecules, cryo sample holder improves the critical dose further if needed. As the ED pattern can easily calculated from the crystalline structure, crystalline form can be identified by comparing the experimental and calculated ED patterns. The ED patterns of L-histidine in orthorhombic and monoclinic forms are shown in Fig. 3 and Fig. 4, respectively. While 13C CPMAS fails to distinguish these two forms (Fig. 1), ED diffraction gives distinct patterns for each crystalline form. This is because the lattice parameters are totally different in these two forms. In addition, ED enables high throughput measurements within one minute compared to PXRD (tens of minutes) and 13C CPMAS (hours). The ED measurements were performed using JEM-ARM200F (JEOL Ltd., Japan). Most TEM instruments are capable of this measurement if high sensitivity camera is installed.
The molecular packing can be investigated using 1H NMR at very fast MAS. 1D 1H NMR spectra of L-histidine (pseudo) polymorphs at 70 kHz MAS are shown in Fig. 5, giving different patterns. Notably, L-histidine in the orthorhombic form (b) gives different pattern from one in the monoclinic form (c). These differences come from different molecular packings. This highlights the advantage of 1H NMR over the 13C CPMAS which fails to identify these two crystalline forms. Thanks to the high abundance (>99%) and Larmor frequency (600 MHz at 14.1 T), it takes less than one minutes to observe these 1D spectra. The difference between two crystalline forms can be magnified by observing 1H/1H homonuclear correlation experiments, which should be sensitive to intermolecular 1H/1H connectivity, thus crystalline packings. The ability of homonuclear correlation is one of the unique features of 1H NMR with the aid of its high sensitivity and abundance. Figure 6 gives 2D 1H double-quantum (DQ) / single-quantum (SQ) homonuclear correlation spectra of L-histidine in orthorhombic and monoclinic forms. While 1D spectra only show the local information, 2D homonuclear correlation spectra also reflect the information on molecular packings, as clearly shown in the overlaid spectra. As the DQ coherences are created through 1H-1H dipolar interactions, the cross peak intensities reflect the spatial proximity. For example, the internuclear distances between H1 and H4 is 4.1 Å for the orthorhombic, it is much shorter and 2.86 Å in the monoclinic form. Although the difference in distance is only 1.4 times, this results in 2.9 times stronger dipolar interactions in the monoclinic form. Thus the H1-H4 correlations are only observed in the monoclinic form. It is 2D experiment, however, the measurement time is typically less than one hour, giving higher throughput than 13C CPMAS.
1H/14N correlation NMR also gives fruitful information. There are multiple possible protonated states in L-histidine. Either or both of the two nitrogens atτand δ on the imidazole ring could be protonated to NH moiety. In addition, L-histidine can be Zwitterion. It is challenging to distinguish these protonated states by either XRD or ED, since both methods are not sensitive to hydrogen positions. However, 1H/14N correlation spectra of L-histidine (Fig. 7) give the clear answer. In the acidic environment in L-histidine·HCl·H2O, three NH correlation appears. This clearly shows bothτand δ nitrogens are protonated. The one more NH correlation appears at low frequency position in the 14N dimension at -260 ppm suggest that this amino acid is Zwitterion with the NH3+ moiety. This is because of small quadrupolar coupling in NH3+ due to the local symmetry. On the other hand, two L-histidine polymorphs only gives two NH peaks for each. It is clear both are Zwitterion and only one ofτand δ nitrogen is protonated. It should be noted that the 1H/14N takes typically less than 10 minutes because both 1H and 14N are abundant nuclei.
All the above measurements were performed using JNM-ECZ600R spectrometer (JEOL RESONANCE Inc., Japan) equipped with 1 mm fast MAS ssNMR probe (JEOL RESONANCE Inc.) at 14.1 T. The sample amount used in the measurements is about 1 mg for each.
Fig.1
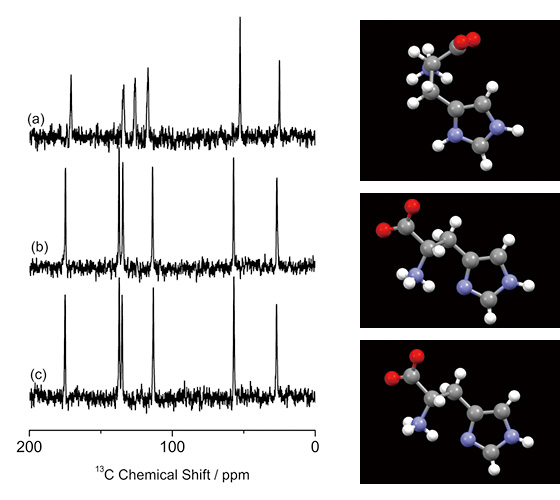
13C CPMAS spectra of (a) L-histidine·HCl·H2O, (b) L-histidine (orthorhombic) and (c) L-histidine (monoclinic), together with the molecular conformations. 256 scans for (a) and 512 scans for (b) and (c) were accumulated. (a) was measured at 16.4 T with a JNM-ECA700II spectrometer (JEOL RESONANCE Inc., Japan). The figure is reproduced from the reference 7.
Fig.2

(a) TEM image and (b) nano-beam (100 nm diameter) diffraction pattern of the microcrystalline L-histidine·HCl·H2O sample. The diffraction pattern was obtained from the area indicated by the white circle in (a). The figure is reproduced from the reference 7.
Fig.3
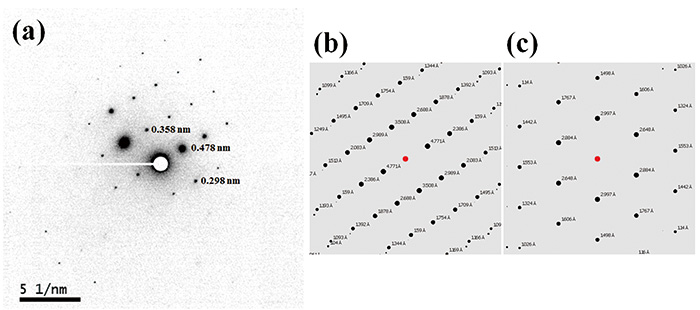
(a) The negatively displayed experimental ED pattern for L-histidine (orthorhombic) and the calculated ED patterns for (b) L-histidine (orthorhombic) and (c) L-histidine (monoclinic). The d-spacings corresponding to the diffraction spots are estimated by the calibrated camera length of 40 cm and a wavelength of 2.51 pm at 200 kV accelerating voltage. The ED pattern was calculated for a [631] zone axis incidence beam. The figure is reproduced from the reference 7.
Fig.4

(a) The negatively displayed experimental ED pattern for L-histidine (monoclinic) and the calculated ED patterns for (b) L-histidine (orthorhombic) and (c) L-histidine (monoclinic). The d-spacings corresponding to the diffraction spots are estimated by the calibrated camera length of 40 cm and a wavelength of 2.51 pm at 200 kV accelerating voltage. The ED pattern was calculated for a [100] zone axis incidence beam. The figure is reproduced from the reference 7.
Fig.5
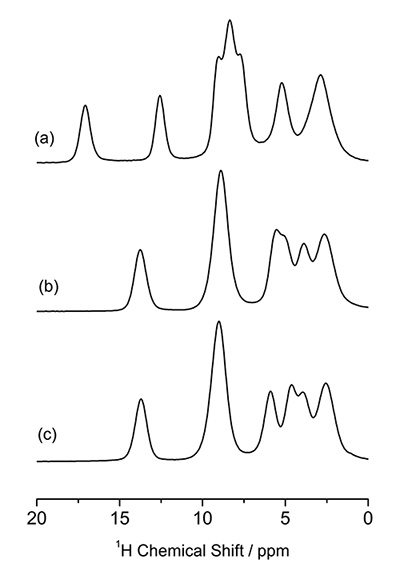
The 1H NMR spectra of (a) L-histidine·HCl·H2O, (b) L-histidine (orthorhombic) and (c) L-histidine (monoclinic). The figure is reproduced from the reference 7.
Fig.6
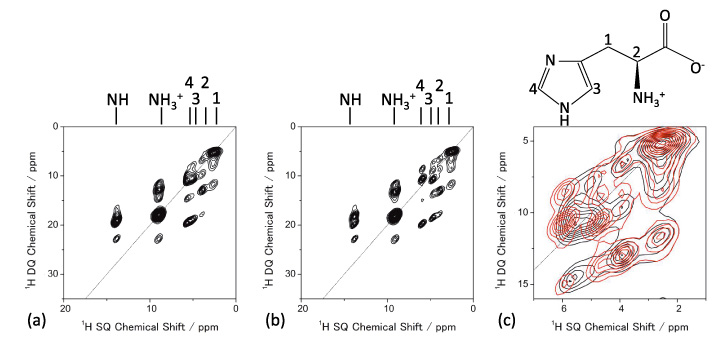
1H DQ/1H SQ NMR spectra of (a) L-histidine (orthorhombic) and (b) L-histidine (monoclinic). The expansion of overlaid spectra is also shown in (c) (L-histidine (orthorhombic) in black and L-histidine (monoclinic) in red). Four scans for each t1 period were accumulated with 32 t1 increments. The figure is reproduced from the reference 7.
Fig.7
The 1H/{14N} NMR spectra of (a) L-histidine·HCl·H2O, (b) L-histidine (orthorhombic) and (c) L-histidine (monoclinic). Spectrum (a) was measured at a 90 kHz MAS rate with a 0.75 mm HXMAS probe (JEOL RESONANCE Inc., Japan). Eight scans for each t1 period were accumulated with (a) 64 and (b)/(c) 32 t1 increments. No 1H–14N recoupling was applied during the mixing time. The figure is reproduced from the reference 7.
New Methods Developed for Crystalline Polymorphs
In spite of the importance of nitrogen in chemistry, pharmacy, material sciences etc., nitrogen NMR is rather limited. This is simply because low sensitivity of 15N NMR due to low abundance of 15N (0.4%). Although 15N is preferred because of its spin quantum number of I = 1/2, the other isotope of 14N is also NMR active nucleus with favorable high abundance of 99%. The integer spin quantum number (I = 1) and presence of quadrupolar coupling hampers the application of 14N NMR. To overcome these problems, we developed the 1H-{14N} heteronuclear multi-quantum coherence (HMQC) measurements at very fast MAS conditions [8]. This method enables high throughput measurements with mass limited samples less than 1 mg. Because of high sensitivity, the method can be expanded into three dimensions for 14N/1H DQ/1H SQ correlation experiments[9]. In addition, the method explores the observation of 35Cl which is often found in pharmaceutical salt [10].
Signal assignments helps to interpret the NMR spectra as discussed above in Fig. 6. While two dimensional homo/heteronuclear correlation helps a lot to assign the signal through two-spin connectivites, three dimensional experiments gives much clearer assignments. To this end, we developed 13C/1H DQ/1H SQ correlation to reveal the local 1H-1H spin network in vicinity of 13C [11]. The complete assignments were achieved from a single three dimensional spectra with 1 mg sample at natural abundance.
Salt/Cocrystal/Continuum[12]
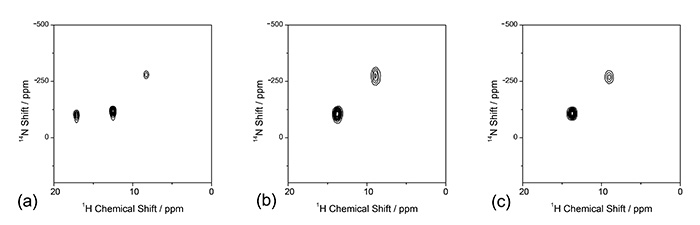
As discussed above, the identification of salt/cocrystal/continuum still remains a big issue in pharmaceutical applications because of lack of ability to locate hydrogen in XRD. The difference between these three classes can be interpreted as the different distances between hydrogen and nitrogen (Fig. 8). Thus, the size of 1H-15N dipolar interactions, which are inversely proportional to cube of the internuclear distances, should give clear answer to salt/cocrystal/continuum.
Here, we propose a combined method of SCXRD and ssNMR. While SCXRD determines global crystalline structure, ssNMR gives local internuclear distances. By combining these two kinds of information, the seamless understanding of the crystalline structure can be established. As a demonstration, first, we synthesize four model multi-component systems shown in Fig. 9. The crystalline structures are determined by SCXRD. The results clearly show the presence of intermolecular contact between N and OH as expected (Fig. 10). The question is whether it is salt/cocrystal/continuum. To answer this question, we tried to determine the hydrogen positions by SCXRD, however, it gives machine dependent values, resulting in unreliable positions. As the crystalline structure determined by SCXRD clearly shows, there is intermolecular 1H/15N coupling. The 1H/15N distances, thus coupling strength between 1H and 15N, should give the answer to salt/cocrystal/continuum problems. For example, 1H/15N distance in SA2 is determined to be 1.25 Å (Fig. 10). With the distances between nitrogen and oxygen of 2.54 Å, which is determined by SCXRD, we can conclude SA2 is continuum as hydrogen is located at the middle of oxygen and nitrogen. It should be noted that SA2 is consisted of strong acid and base with ΔpKa larger than 3. This result highlights the importance of SCXRD/ssNMR measurements to identify salt/cocrystal/continuum even for the system with large ΔpKa. The structures and internuclear distances of the other three systems have been successfully determined (Fig. 10). The bottleneck of this method is experimental time for 1H-15N distance measurement. In addition to the low natural abundance of 15N and small 1H-15N couplings, these pharmaceutical samples typically show very long 1H T1 relaxation time. Moreover, the need of fast MAS reduces the sample volume, resulting in further sensitivity reduction. We have developed a method to improve the throughput as discussed in the following section, each measurements still requires 4-5 days. We agree with the fact of the limited throughput. Nevertheless, we believe this method is quite useful as no other method gives clear answer.
All the NMR measurements were conducted with JNM-ECA700II NMR spectrometer (JEOL RESONANCE Inc.) using 1 mm double resonance fast MAS probe (JEOL RESONANCE Inc.) at 16.4 T.
Fig.8
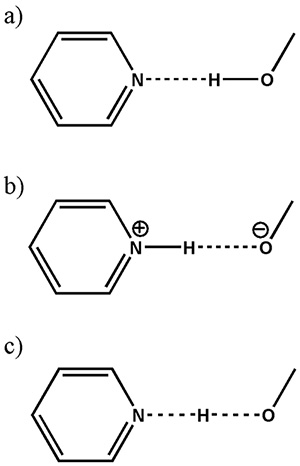
Schematic presentation of (a) a cocrystal, (b) a salt and (c) a continuum (where the H-atom position is shared between the two heavy atoms) in a typical O…H…N interaction. The figure is reproduced from the reference 12.
Fig.9
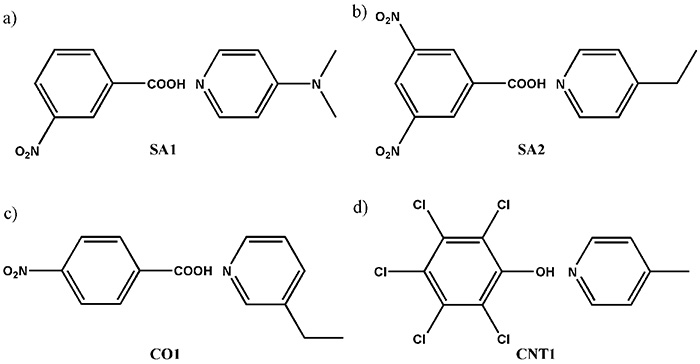
Schematic representation of the compounds used in the present study, showing (a) SA1 (3-nitrobenzoic acid and N,N-dimethypyridin-4-amine), (b) SA2 (3,5-dinitrobenzoic acid and 4-ethylpyridine), (c) CO1 (4-nitrobenzoic acid and 3-ethylpyridine) and (d) CNT1 (pentachlorophenol and 4-methylpyridine). The figure is reproduced from the reference 12.
Fig.10
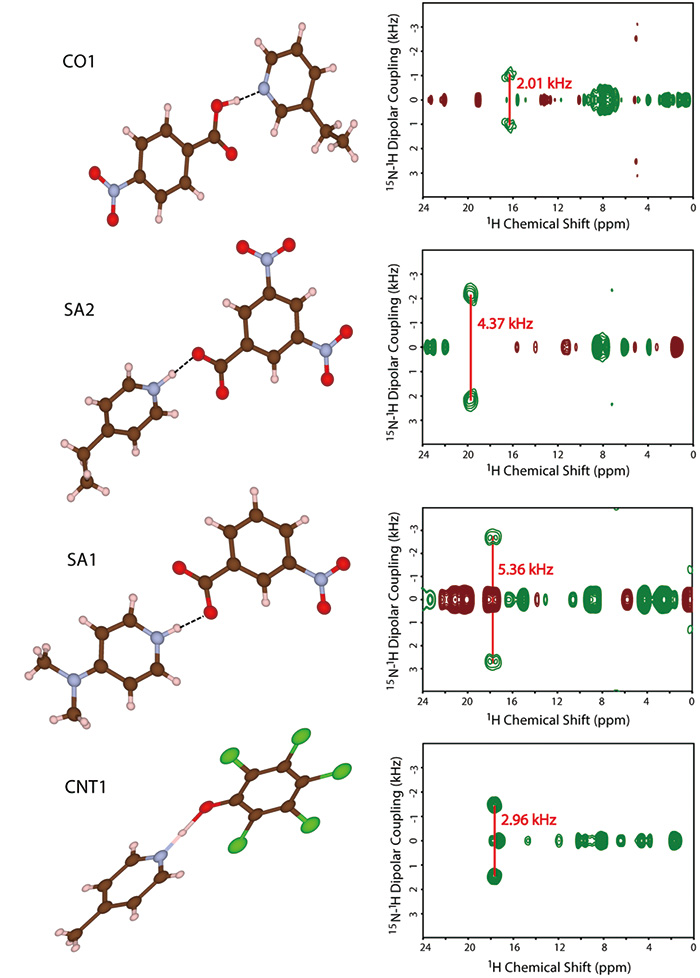
The molecular structures and the two-dimensional invCP-VC spectra (15N-1H dipolar couplings versus 1H chemical shift) of SA1, SA2, CO1 and CNT1. The figure is reproduced from the reference 12.
New Methods Developed for Salt/Cocrystal/Continuum
The size of 1H/15N dipolar coupling should be precisely measured. However, it is not straightforward task because of 1) low abundance of 15N (0.4%), 2) small gyromagnetic ratio (1/10 of 1H), and 3) abundant 1H nuclei which are not bonded to 15N, resulting in low sensitivity, small dipolar coupling (1-7 kHz), and overlapping of 1H resonances, respectively. More importantly, the previous methods tend to show the 1H/15N dipolar coupling which depends on experimental conditions, giving unreliable distances. To overcome these difficulties, we have introduced inverse detected CP with variable contact time (inv CP-VC) method at fast MAS [13]. Firstly, the method gives well defined dipolar splittings which is independent to experimental conditions [14]. Actually, the scaling factor of inv CP-VC method largely depends on the rf field strength. However, signals from the area with B1 inhomogeneity are scattered over the broad spectral range, making these signals invisible. Thus the signals from homogeneous B1 field are selectively observed. The sensitivity enhancement by 1H detection in inv CP-VC alleviates the low sensitivity due to small abundance and sample volume. Experimentally, it is shown that inv CP-VC is capable to monitor a 1H/15N coupling of 2 kHz. The inv CP-VC works as filtering of 1H resonances, giving selective observation of 1H in vicinity of 15N. This avoids the complexity of overlapped resonances between 1Hs. In addition, inv CP-VC paves a way to analyze mass limited samples less than 1 mg.
Repetition delay must be inserted between the consecutive scans in NMR measurements so that the spin system returns to close to the thermal equilibrium before starting the NMR measurements. As the repetition delay should be an order of T1 relaxation time, which is typically much longer than the rest of the experiments, T1 relaxation time is the dominating factor of experiment time in NMR. In the inv CP-VC experiments, the magnetization starts from 1H, thus, 1H T1 is important. However, well crystalline low-molecular weight APIs tend to have very long 1H T1 relaxation time of tens to hundreds of seconds, reducing the throughput dramatically. At the fast MAS rate, the issue is more severe, because 1H-1H spin diffusion is suppressed, resulting in longer 1H T1 relaxation time. Since NH protons, which are of our interest, are spatially and spectrally isolated from the other 1Hs, very slow 1H-1H spin diffusion hampers the 1H magnetization from recovering to its thermal equilibrium. This results in slower 1H T1 relaxation time of NH protons compared to the rest of 1Hs. To overcome this difficulty, we have applied radio-frequency driven recoupling (RFDR) sequences on 1H, during repetition delay [15]. This enhances the 1H-1H spin diffusion, bringing the magnetization from rapidly relaxing 1H to NH protons. Since the relaxation mechanisms of rapidly relaxing proton are utilized multiple times, not only NH proton but also the overall sensitivity could also be improved. We also optimized the phase cycling in the RFDR sequence suitable to 1H mixing [16, 17].
Conclusion
Both TEM and NMR are continuously improved even now despite the fact that they have been introduced more than 70 years ago. The recently introduced high sensitivity camera in TEM opens a way to the novel application in cryo TEM to determine the protein structure using the single particle analysis approach. The sensitivity enhancement is beneficial for not only protein but also beam-sensitive samples including low-molecular weight API. Another big burst is observed in fast MAS technology of ssNMR, which enables 1H NMR even rigid solids. These new developments not only provide the new information which had previously been inaccessible but also improve the throughput, which is one of the key factors for wide spread of use. In addition, it is quite important to apply several different analytical methods which are complimentary to each other. The proper combination provides useful information with high throughput. Indeed, JEOL Ltd. also pushes the YOKOGUSHI strategy to offer attractive combinations of equipments.
In this article, we combine XRD, ED, and ssNMR to solve the structural problem in pharmaceutical sciences including crystalline polymorphs and salt/cocrystal/continuum issues. For the crystalline polymorphs issue, we demonstrate the combined approach of ED and 1H fast MAS NMR to identify the crystalline form, as a method complimentary to the PXRD and 13C CPMAS. ED enables crystalline form determination from nano- to micro-sized single crystals. On the other hand, 1H fast MAS can distinguish the crystalline form with similar molecular conformation. In addition, fast MAS enables 1H/14N correlations, giving proton positions close to nitrogen nuclei. To identify the salt/cocrystal/continuum, the combined approach of SCXRD and ssNMR at very fast MAS rate is introduced. While SCXRD provide the global molecular structure, it fails to determine the proton positions which are crucial to salt/cocrystal/continuum issue. On the other hand, ssNMR gives precise 1H-15N distances, although the global structure is hardly obtained. This approach provides seamless understanding of molecular structure from crystalline structure to hydrogen bonding to answer salt/cocrystal/continuum issue. The formation of continuum in the multi-component systems with ΔpKa > 3 highlights the importance of thorough investigation using SCXRD and ssNMR.
Acknowledgments
All the works presented here were performed together with our group members at RIKEN CLST-JEOL collaboration laboratory. I would like to thank to the all the group members and to our colleague at JEOL Ltd. and JEOL RESONANCE Inc. for great support. I also would like to acknowledge our friends at Indian institute of sciences at Bangalore, AMES lab, Kyoto University, Tokyo University of Agriculture and Technology, University of Warwick, University of Lille, University of Michigan for fruitful collaborations.
References
- R. K. Harris, J. Pharm. Pharmacol. 59 (2007) 225–239.
- M. Geppi, G. Mollica, S. Borsacchi, C. A. Veracini, Appl. Spectrosc. Rev. 43 (2008) 202–302.
- F. G. Vogt, Future Med. Chem. 2 (2010) 915–921.
- Y. Nishiyama*, Fast Magic-Angle Sample Spinning Solid-State NMR at 60-100 kHz for Natural Abundance Samples, Solid State Nuclear Magnetic Resonance, 78 (2016) 24-36. DOI: 10. 1016/j. ssnmr. 2016. 06. 002.
- Y. Nishiyama*, Solid-state NMR under ultrafast MAS rate of 40 ‒ 120 kHz, in Experimental Approaches of NMR Spectroscopy ‐ Methodology and Application to Life Science and Materials Science, Springer (2017).
- T. Kobayashi, Y. Nishiyama*, M. Pruski*, Heteronuclear correlation spectroscopy with inverse detection, in Modern Methods in Solid-State NMR: A Practitionerʼs Guide, Royal Society of Chemistry (2018).
- T. Oikawa, M. Okumura, T. Kimura, Y. Nishiyama*, Solid-state NMR meets electron diffraction: Determination of crystalline polymorphs of small rganic microcrystalline samples, Acta Cryst. C73 (2017) 219‒228. DOI: 10. 1107/S2053229617003084.
- Y. Nishiyama*, Y. Endo, T. Nemoto, H. Utsumi, K. Yamauchi,
Are you a medical professional or personnel engaged in medical care?
No
Please be reminded that these pages are not intended to provide the general public with information about the products.
Contacts
JEOL provides a variety of support services to ensure that our customers can use our products with peace of mind.
Please feel free to contact us.